24 февраля 2020
Клиническая характеристика болезни моямоя у пациентов с синдромом Дауна
Инсульт – острое нарушении функции головного мозга, вызванное ишемией или кровоизлиянием.
Транзиторная ишемическая атака (ТИА) – обратимое нарушение церебральных функций, обусловленных ишемией, при котором симптомы сохраняются менее суток. ТИА – важный признак угрозы инсульта.
Ишемические инсульты являются серьезной проблемой не только для взрослых пациентов. Частота их среди детей составляет 1,2 на 100 000 в год [1]. Около 3 % случаев инсульта заканчивается летально, а у 74 % больных впоследствии проявляется серьезный неврологический дефицит [2]. Несмотря на небольшую частоту инсультов у детей, каждый случай требует тщательного анализа, чтобы не допустить повторных эпизодов ишемических атак и предотвратить появление тяжелых неврологических нарушений.
У детей острые ишемические состояния часто могут быть обусловлены заболеваниями сердца и транзиторными артериопатиями. Однако от 3 до 10 % случаев инсультов головного мозга вызваны болезнью моямоя (БММ) [3, 4].
БММ – редкое хроническое заболевание сосудов головного мозга, для которого характерно медленное (на протяжении месяцев и даже лет) прогрессирующее сужение просвета внутричерепных сегментов внутренних сонных артерий и сосудов виллизиева круга. Важная отличительная черта этого заболевания – формирование сети коллатеральных сообщений, что на ангиограммах создает впечатление легкой дымки. Именно эта особенность определила название болезни: «моямоя» в переводе с японского – клубок дыма. Первое описание БММ относится к 1957 году и принадлежит Такеуши (Takeuchi) и Шимидзу (Shimizu) [5].
Лечение БММ хирургическое, медикаментозная терапия неэффективна [6]. Цель лечения заключается в восстановлении кровоснабжения сосудов головного мозга.
Ранее эту болезнь рассматривали исключительно как самостоятельное заболевание. Позднее была отмечена повышенная частота ассоциации БММ с различными наследственными и системными нарушениями.
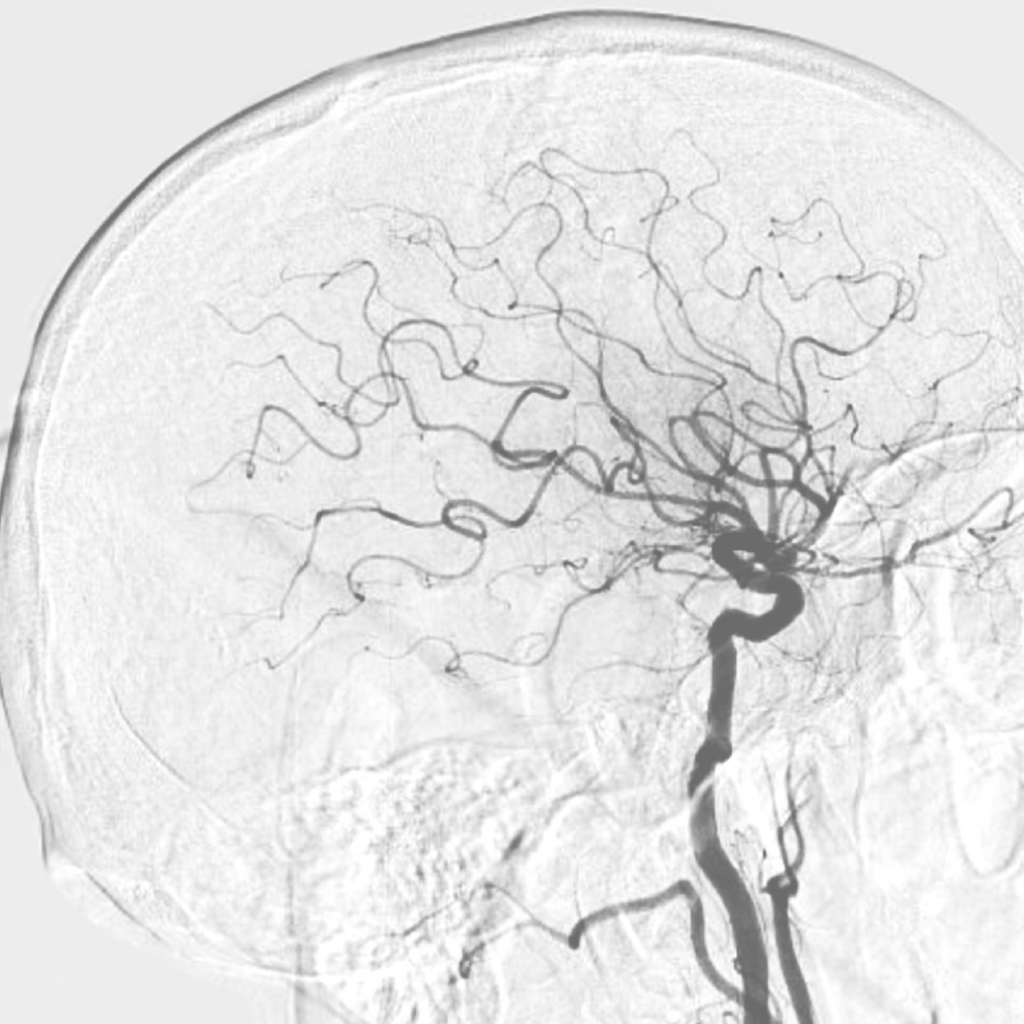
В последние годы появились исследования, указывающие на повышенную частоту этого редкого заболевания у детей с синдромом Дауна (СД). Всего в литературе встречается описание около 80 случаев пациентов с БММ и трисомией по 21-й паре хромосом. В 2015 году была опубликована работа, в которой представлен опыт лечения 32 пациентов с синдромом Дауна и БММ в детской больнице Бостона за период с 1985 по 2012 год [7]. Это исследование заслуживает особого внимания, поскольку авторы провели сравнение клинического течения БММ и результатов оперативного лечения у детей с синдромом Дауна и без него.
Всего за указанный период в клинике Бостона получили хирургическое лечение по поводу БММ 500 больных. Пациентов с БММ и СД было 32 (15 девочек и 17 мальчиков), что составило 6,4 %. Их возраст на момент лечения – от 1,8 до 29,3 года; 29 из 32 больных с СД – это пациенты до 21 года. Интересной особенностью оказалась высокая частота врожденных пороков сердца в этой группе. У 24 из 32 пациентов (75 %) были выявлены различные пороки сердца, в том числе дефекты межжелудочковой перегородки, тетрада Фалло и др. (в то время как частота врожденных пороков сердца у детей с трисомией-21 в целом не превышает 40 %). Дебют проявлений БММ у детей с СД, в сравнении с детьми без синдрома, более поздний, в среднем в 8,4 года. У детей без синдрома средний возраст появления клинических симптомов БММ составил 6,5 лет. При этом у 100 % детей с СД на ангиограмме были характерные симптомы болезни. У пациентов без синдрома характерные признаки были отмечены только в 75 % случаев.
Среди других клинических особенностей БММ у детей с трисомией-21 отмечена более высокая частота инсультов: у 27 из 32 больных (87 %). ТИА встречались реже, только у 14 больных (42 %). Более распространен и судорожный синдром у детей с СД, вошедших в исследуемую группу. Судороги были диагностированы у 8 детей (26 %). А среди детей без синдрома судороги встречались менее чем в 10 % случаев.
Хирургическое лечение БММ у пациентов с СД также имеет некоторые особенности. Прежде всего, при выборе тактики анестезиологического пособия необходимо учитывать наличие сопутствующей патологии (пороки сердца, гипотиреоз, ЛОР-патологию и др.).
С одной стороны, течение БММ у детей с СД имеет более выраженные черты (преобладают инсульты, чаще встречается судорожный синдром). С другой – результаты хирургического лечения, как оказалось, у них лучше. Интересно, что у пациентов с СД отмечался стойкий клинический и радиологический эффект. Отдаленные результаты хирургического лечения детей с трисомией по 21-й паре хромосом превосходят таковые у пациентов без СД. Повторные инсульты возникали у пациентов с СД нечасто и не ранее чем через 7,5 лет.
Таким образом, своевременная диагностика БММ у детей является важнейшим звеном в выборе эффективной тактики лечения, а также профилактики повторных ишемических атак головного мозга.
Рассмотрим в качестве примера клинический случай БММ у ребенка с СД с манифестацией на первом году жизни.
Доношенная девочка 3 месяцев жизни. Пренатально, на сроке 30–31 неделя, на УЗИ выявлен порок сердца, предположен синдром Дауна. Роды вторые, в 39 недель, самостоятельные. При рождении масса девочки – 2630 г, длина – 45 см, оценка по шкале АПГАР – 7/8 баллов. В связи с особенностями фенотипа, характерными для синдрома Дауна, взята кровь для хромосомного исследования, кариотип: 47, ХХ, +21 – регулярная трисомия 21-й хромосомы. Ребенок переведен в отделение патологии новорожденных, где находился с диагнозом «Врожденный порок сердца: общий открытый атриовентрикулярный канал, тип А по Растелли. Вторичный дефект межпредсердной перегородки (7 мм). Легочная гипертензия. Открытый артериальный проток (3 мм). Внутриутробная инфекция без очага. Гипоксически-ишемическое поражение центральной нервной системы, синдром угнетения. Постгипоксическая нефропатия. Внутриутробная гипотрофия 1–2 степени. Синдром Дауна». В возрасте 2,5 месяцев проведена операция суживания легочной артерии. В ходе госпитализации выявлен субклинический гипотиреоз, назначена заместительная терапия. Выписана спустя 10 дней после лечения. На 2-е сутки после выписки поступила в детское инфекционное отделение ДГКБ № 9 им. Сперанского с жалобами на слабость, отказ от еды. Состояние ребенка тяжелое за счет признаков инфекционного токсикоза, на фоне дыхательной недостаточности у ребенка с врожденным пороком сердца, синдромом Дауна. В неврологическом статусе отмечалась мышечная гипотония, клонусы правой руки при осмотре. В клиническом анализе крови при поступлении выявлен тромбоцитоз до 589 тыс/мкл (норма:149–409), в биохимическом анализе крови повышение С-реактивного белка до 13 мг/л (норма: 0,1–8,2), остальные показатели в норме. Тиреоидный статус в норме. В коагулограмме: снижение АЧТВ до 19,1 сек (норма 25,4–36,9), с нормальным уровнем фибриногена – 2,25 г/л (норма 1,41–4,37), повышением протромбинового времени до 13,1 сек (9,4–12,1), МНО до 1,22 INR (норма 0,86–1,22) и тромбинового времени до 16,8 сек (10,3–16,6). По данным МРТ головного мозга в сосудистом режиме выявлено острое нарушение мозгового кровообращения – картина, характерная для БММ. В динамике состояние ребенка улучшилось. Выписана под наблюдение кардиолога и невролога.
Безусловно, возраст нашей пациентки значительно отличается от среднего возраста начала БММ у описанных ранее пациентов. Однако работы, указывающие на связь между СД и БММ, появились относительно недавно и, возможно, у некоторых детей первых лет жизни БММ не была диагностирована. К тому же в качестве наиболее вероятной причины острого нарушения мозгового кровообращения у ребенка в нашем случае без проведения МРТ головного мозга можно было бы предположить тромбоз сосудов головного мозга, возникший после оперативного лечения порока сердца. А это, в свою очередь, принципиально меняет лечебную тактику.
Более того, в последнее время значительно переоценивается вклад наследственных тромбофилий в этиологию ишемических состояний головного мозга у детей. Все это, безусловно, откладывает постановку правильного диагноза и начало эффективного лечения этого прогрессирующего заболевания.
Цель данной статьи – обратить внимание на редкое заболевание, при котором, однако, имеется лечение, способное предотвратить развитие тяжелого неврологического дефицита. Очевидно, что все случаи ишемических атак и инсультов у детей требуют проведения МРТ головного мозга. В последнее время все больше исследователей рекомендуют рассматривать БММ как одну из основных причин инсультов и ТИА у больных с СД. Более того, большинство зарубежных врачей склоняются к целесообразности проведения скрининга БММ у детей с СД [7–11]. Эта рекомендация пока не получила широкого распространения, поскольку продолжается накопление клинических данных и изучение этой болезни у пациентов с СД.
Литература
1. Discovery of asymptomatic moyamoya arteriopathy in pediatric syndromic populations: radiographic and clinicalprogression / N. Lin et al. // Neurosurgical Focus. 2011. Vol. 31, Iss. 6. Р. E6.
2. Scott R. M., Smith E. R. Moyamoya disease and moyamoya syndrome // The New England journal of medicine. 2009. Vol. 360. Р. 1226–1237.
3. Moyamoya syndrome associated with Down syndrome: outcome after surgical revascularization / A. Jea et al. // Pediatrics. 2005. Vol. 116. Р. e694–e701.
4. Fallon P., Moorthy B., Ganesan V. Presumed perinatal stroke in a child with Down syndrome and moyamoya disease / K. Pysden et al. // Developmental medicine & Child neurology. 2010. Vol. 52. Р. 212–214.
5. Takeuchi K., Shimizu K. Hypoplasia of the bilateral internal carotid arteries // Brain Nerve. 1957. Vol. 9. Р. 37–43.
6. Yamada I., Matsushima Y., Suzuki S. Childhood moyamoya disease before and after encephalo-duro-arterio-synangiosis: an angiographic study // Neuroradiology. 1992. Vol. 34. Р. 318–322.
7. Down syndrome and moyamoya: clinical presentation and surgical management / A. P. See et al. // Journal of Neurosurgery: Pediatrics. 2015. Vol. 16. Р. 58–63.
8. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events / S. R. Dobson et al. // Blood. 2002. Vol. 99. Р. 3144–3150.
9. Surgical treatment of moyamoya syndrome in patients with sickle cell anemia: outcome following encephaloduroarteriosynangiosis / T. C. Hankinson et al. // Journal of Neurosurgery: Pediatrics. 2008. Vol. 1. Р. 211–216.
10. Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young / E. S. Roach et al. // Stroke. 2008. Vol. 39. Р. 2644–2691.
11. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome / E. R. Smith et al. // Neurosurgical Focus. 2009. Vol. 26, Iss. 4. P. E10.
Автор:
Благотворительный фонд «Даунсайд Ап» / Семенова Наталия Александровна / Митина Ю. Ю. / Паллак Ж. Ю.
Возраст до:
18
Источник:
Журнал «Синдром Дауна. XXI век» № 2 (23)
Категория:
Родителям / О синдроме Дауна
Тип материала:
Статьи
Теги материала:
медицинское сопровождение / сопутствующие заболевания / мозг / лечение
Количество показов:
6939